糖尿病でインスリンが出にくくなる新たな原因を解明
群馬大学生体調節研究所の、井上亮太助教、白川純教授らの研究グループは、横浜市立大学、アルバータ大学(カナダ)、理化学研究所、ジョスリン糖尿病センター(米国)等との共同研究で、糖尿病の膵β細胞からのインスリン分泌が低下する新たな原因を解明しました。
糖尿病は膵臓の膵島に存在するβ細胞から分泌されるインスリンが減少することで発症します。また、糖尿病では、過剰な糖(グルコース)が膵β細胞からのインスリン分泌低下を助長し、さらなる血糖上昇を招きますが、その原因は充分に解明されていません。
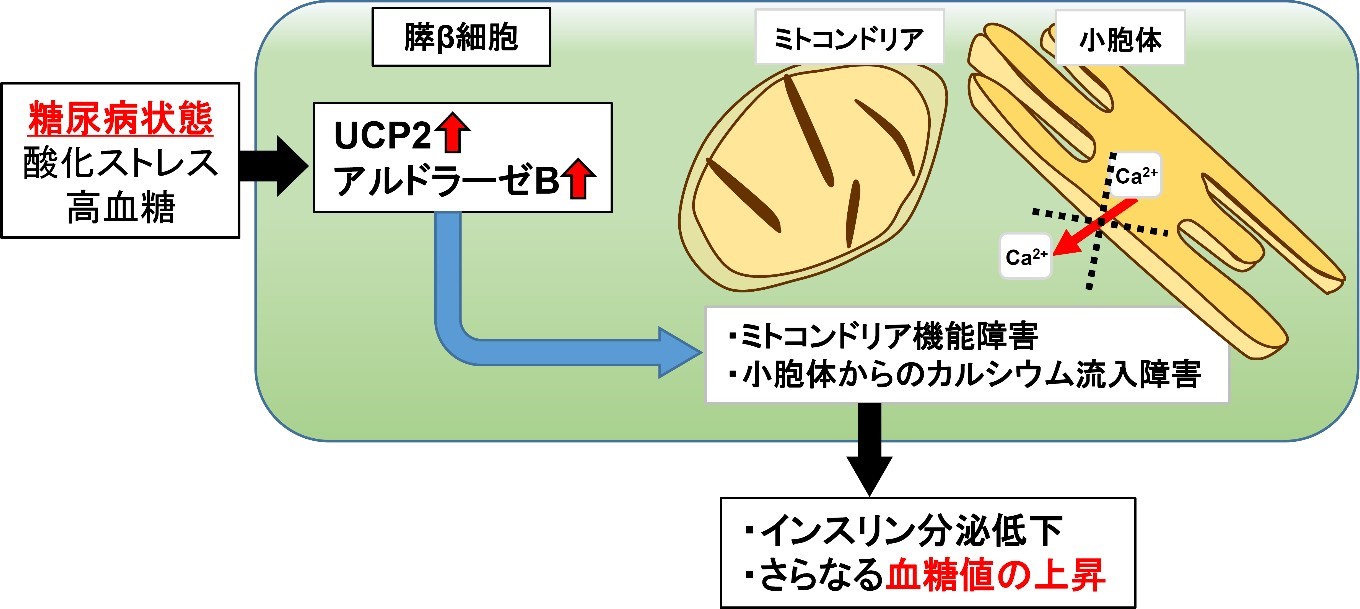
今回の共同研究で、糖尿病ドナー由来の膵島で増えているUCP2 (Uncoupling protein 2) というミトコンドリアの蛋白に着目しました。ミトコンドリアでは、細胞が活動するために必要なATPという物質が生成されます。UCPはATPを産生するためのエネルギーを熱へ変換する役割(脱共役)を持ちます。UCPは余計なエネルギーを熱に変換するので、肥満になりにくくなる蛋白であると考えられています。糖尿病でなぜUCP2が増えているのかは不明でした。そこで、膵β細胞でUCP2が過剰に作られる遺伝子組換えマウス(βUCP2Tgマウス)を作成し、UCP2の膵β細胞での役割を解析しました。
βUCP2Tgマウスは、高血糖とインスリンの分泌が減りヒトの糖尿病に似た病態を示しました。βUCP2Tg マウスの膵島では、ミトコンドリアでのインスリン分泌に重要なATP産生と小胞体からのカルシウム放出に異常があり、これらがインスリン分泌減少の原因であると考えられました。βUCP2Tgマウスの膵島における脱共役は糖尿病がないマウスと同等であり、UCP2は脱共役以外の作用でインスリン分泌低下を引き起こす可能性が示唆されました。さらなる解析により、UCP2がアルドラーゼBという酵素を増やすことを見出しました。
膵β細胞でアルドラーゼBが増えると、UCP2が増えたときと同様に、インスリン分泌低下、ミトコンドリア機能異常および小胞体からのカルシウム放出障害が生じることが分かりました。さらに、アルドラーゼBの遺伝子発現を抑制することで、UCP2が増えたことによるインスリン分泌低下が回復しました。
本研究により、糖尿病の膵β細胞で増加するUCP2が、アルドラーゼBを介してミトコンドリア機能異常および小胞体からのカルシウム放出障害を誘導し、膵β細胞からのインスリン分泌低下を引き起こすことで血糖値の更なる上昇を招く新たな機構が明らかとなりました。また、本研究により、UCP2もしくはアルドラーゼB を標的としたインスリン分泌を回復させる糖尿病治療への展開が期待されます。本研究の成果は2022 年7月15日号のiScience誌(Cell press 社:米国)に掲載されました。
本件のポイント
- 2型糖尿病患者の膵島で増えているUCP2の役割は不明であった。
- UCP2は脱共役を介さずに、膵β細胞から分泌されるインスリンを低下させた。
- UCP2の過剰発現によりミトコンドリア機能異常、小胞体からのカルシウム流入異常が生じた。
- UCP2はアルドラーゼBという酵素を増やしインスリンの分泌を低下させた。
- アルドラーゼBを抑制することにより、UCP2によるインスリン分泌低下が回復した。
本件の概要
2型糖尿病は膵β細胞からのインスリン分泌低下と肥満などによるインスリン抵抗性により発症します。本国の糖尿病患者数は約2000万人と多く、腎症による人工透析患者数の増加などが問題となっています。糖尿病状態では、高血糖や炎症などにより膵β細胞からのインスリン分泌が低下し、さらに血糖値が上昇することが分かっていますが、その原因は充分に分かっていません。糖尿病患者で減少する膵β細胞からのインスリン分泌を回復させることが、糖尿病の治療に繋がると考えられます。
我々は先行研究において、高グルコース刺激によりマウス膵島で遺伝子発現が増える蛋白として として、ミトコンドリアのUncoupling protein 2 (UCP2) を見つけました。UCP2は糖尿病ドナー由来の膵島で増えていることが分かっていますが、膵β細胞でなぜUCP2が増えるのかは不明で した。UCP2と構造が似たUCP1は、ミトコンドリアが豊富な褐色脂肪に存在し、細胞のエネルギー源となるATPの産生を減らし、そのエネルギーを熱に変換します(脱共役)。膵β細胞においては、UCP1ではなくUCP2が発現しています。
今回我々は、膵β細胞でUCP2が過剰に作られる遺伝子改変マウス(βUCP2Tgマウス)を作成し、膵β細胞でのUCP2の役割を検討しました。βUCP2Tgマウスは、高血糖とインスリンの分泌が減りヒトの糖尿病に似た病態を示しました。膵β細胞でUCP2が増えると、インスリン分泌に重要なATPは減少しましたが、脱共役は糖尿病がないマウスと同等でした。さらなる解析により、UCP2がアルドラーゼBという解糖系酵素を増やすことを見出しました。アルドラーゼBは主に肝臓に存在し、果糖の代謝に関わることが知られていますが、膵β細胞での役割は不明です。膵β細胞においてアルドラーゼBが増えると、UCP2が増えた時と同様に、ミトコンドリア機能異常、インスリン分泌減少が生じました。また、膵β細胞からのインスリン分泌は、カルシウムにより調節されることが知られています。UCP2やアルドラーゼBが増えると、小胞体というカルシウムを貯蓄する細胞内小器官から細胞質へのカルシウム供給が減ることが分かりました。これらのミトコンドリア機能低下やカルシウム供給異常は、アルドラーゼBを抑制することにより回復しました。
さらに、本研究室ではヒト膵島を用いた検討が可能であり、上記の動物モデルで認めたUCP2によるインスリン分泌の減少が、ヒト膵島においても観察されることを確認しました。
以上より、糖尿病の膵β細胞で増えるUCP2が、アルドラーゼBを介してミトコンドリア機能低下および小胞体からのカルシウム供給異常を誘導し、膵β細胞からのインスリン分泌低下を引き起こすことで血糖値の更なる上昇を招く新たな機構が明らかとなりました。また、本研究によりUCP2の脱共役とは独立した新たな機能が示され、UCP2もしくはアルドラーゼBを標的としたインスリン分泌を回復させる糖尿病治療への展開が期待されます。
本研究は、国立研究開発法人日本医療研究開発機構(AMED)、科学研究費助成事業、および民間助成金からの助成に加え、1型糖尿病の患者及び家族による認定NPO法人であるIDDMネットワークの支援を受けて行われました。
関連リンク
論文詳細
- 論文名:Uncoupling protein 2 and aldolase B impact insulin release by modulating mitochondrial function and Ca2+ release from the ER
- 論文著者:井上亮太1,2, 都野貴寛1,2, 富樫優2, 奥山朋子2, 佐藤葵1, 西山邦幸1,2, 京原麻由2, Li Jinghe1,2, 福島説子1, 金達也3, 宮下大介2, 紫葉裕介2, 跡部好敏4, 清成寛5, 坂東可菜5, A.M. James Shapiro3, 船越健悟4, Rohit N. Kulkarni6, 寺内康夫2, 白川 純1, 2,* (1. 群馬大学生体調節研究所代謝疾患医科学分野、2. 横浜市立大学医学部分子内分泌?糖尿病内科、3. アルバータ大学臨床膵島研究室、4. 横浜市立大学医学部神経解剖学、5. 理化学研究所 生命機能科学研究センター、6. ハーバード大学ジョスリン糖尿病センター. *, 責任著者)
- iScience誌(Cell Press:米国)
- 公開日:2022年7月15日
本件に関するお問合せ先
群馬大学? 生体調節研究所? 代謝疾患医科学分野? 教授? 白川? 純
TEL:027-220-8850
E-MAIL:jshira★gunma-u.ac.jp
群馬大学 生体調節研究所庶務係 係長 富澤 一未
TEL:027-220-8822
E-MAIL:kk-msomu4★ml.gunma-u.ac.jp
★を半角の@に変更してください